Abraham Herzog-Arbeitman, Department of Chemistry, University of Chicago, Chicago, United States
Polymeric materials, which include plastics, fabrics and the vast majority of biological substances, are abundant in modern society. On the molecular scale, these materials are built from the interconnection of similar chemical subunits to form chainlike macromolecules. The medical, electrochemical, synthetic and materials science communities have increasingly turned their attention to polyelectrolytes, a subset of polymers with electric charge, in part because of their ability to spontaneously and reversibly phase-separate (complex) in aqueous solution. This entropic and electrostatic separation can yield well-defined nanoparticles, dynamic gels and other useful materials. These materials are called polyelectrolyte complexes (PECs) and have formed the basis of complex-core micelle technologies that show promise as nanoscale drug-delivery vehicles for cancer treatment, and which have long been suspected to play a role in the chemical origin of life. This article describes the thermodynamics underlying polyelectrolyte complexation, and then illustrates the application of PECs in medicine and their possible role in the beginning of life, interconnecting these far-flung domains through their reliance on the spontaneous organisation of nanoscale space by chemical activity, the singular physical process of electrolyte complexation.
Keywords: Polyelectrolyte, thermodynamics, drug-delivery, origin of life, micelle, complexation
Polymers are long, chainlike molecules built from the interconnection of repeating subunits; the word 'polymer' comes from the Greek polús, meaning 'many', and méros, which is 'part'. From a synthetic perspective, polymers are straightforward to create because the links between monomers (the subunit of choice) are essentially identical – a single reaction that joins two monomers can often join ten thousand. Accordingly, polymeric compounds are ubiquitous: walk into a room, and anything that is not metal or stone is probably polymer, including people – the protein, nucleic acid and carbohydrate building blocks that make up living things are all polymeric. A special class of electric charge-containing polymers, labelled polyelectrolytes, has received increasing attention, in part because of their ability to form reversible polymer-rich phases in solution called polyelectrolyte complexes, or PECs.
Associations between the polymer chains within the PEC phase are electrostatic, meaning that crosslinks are not chemical, and can be fully reversed. PECs are therefore often stimuli-responsive (Tekaat et al., 2015; Krogstad et al., 2014b), self-healing (Zhang et al., 2016) and self-assembling (Overbeek and Voorn, 2005). The chemical structure of the constituent polyelectrolytes largely determines the mechanical and physical properties of the PEC phase (Ting et al., 2018). Because of their modular construction, highly tuneable properties and unusual abilities, PECs are of interest in chemistry as nanoscale reaction vessels, and in medicine as bioadhesives, biomolecule sensors and nanoparticle drug-delivery vehicles, potentially including CRISPR-Cas9 technologies (Ramasamy et al., 2016; Guvendiren et al., 2012; Zhao et al., 2016). Furthermore, PECs are model systems of interest in fields concerning chemical autopoiesis and the origin of life (Oparin, 1953). Elucidating the nanoscale design principles that relate polyelectrolyte structure to complex architecture in solution is crucial for each of these fields and materials science in general, and generates strong continued interest in polyelectrolyte complexation.
This article discusses the basic thermodynamic principles behind polyelectrolyte complexation and shows how these principles generate behaviour relevant to drug-delivery nanovehicles and chemical autopoiesis. While this article is not a comprehensive literature review of the wide and deep subfields of polyelectrolyte physics and engineering, the author hopes to generate interest among the general scientific readership and supply an entry point into this fascinating and interdisciplinary literature. The aforementioned applications of polyelectrolyte research are chosen to demonstrate the power of polyelectrolyte complexation to link far-flung disciplines by the underlying chemistry and physics examined here, which relates to the spontaneous organisation of structure, activity and information on the nano- to micro-scale, and the general control thereof through thermodynamic principles.
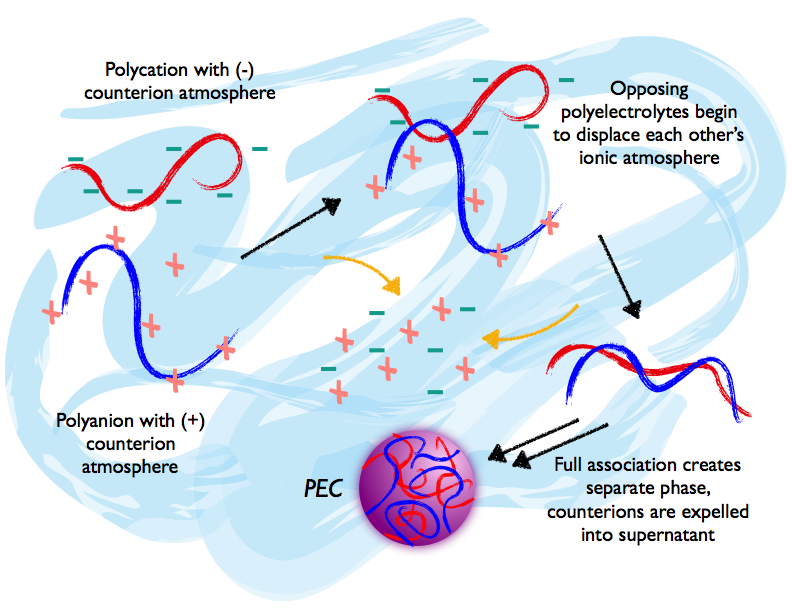
Polyelectrolyte complexes are formed when oppositely charged polymers (polyanions containing negative charges and polycations containing positive charges) are mixed in solution, which is generally aqueous. The charges on the polycations and polyanions attract each other so that the polymers are bound electrostatically, forming a separate phase apart from the rest of the solution (Krogstad et al., 2014a). The strength of these electrostatic interactions and the structure of the monomer segments involved determine whether this phase is liquid-like (termed a coacervate) or solid-like (sometimes called a non-equilibrium complex or simply a PEC) (Krogstad et al., 2014b). Counterions included with the polyions for electroneutrality are expelled into the bulk aqueous solution (the supernatant) (Li et al., 2018). This complexation interaction in aqueous media occurs rapidly without external stimulus, constituting the spontaneous self-assembly of the PEC phase.
Self-assembly is a rare ability among synthetic materials, having developed only recently in the field of nanotechnology. Nature, however, has mastered this technique to turn cleverly arranged simple molecules into increasingly elaborate designs, creating the complex and dynamic molecular behaviour that is required for life as we know it. For example, protein folding is generally regarded as a self-assembly process directed by the sequence of amino acid residues in polypeptide primary structure, despite the role of chaperone proteins becoming increasingly recognised (Balchin et al., 2016). Each amino acid has simple chemical and physical activity (e.g. hydrophobicity, acidity, metal affinity, steric bulkiness, etc.) but when arranged in a certain order, these functional groups interact to fold the protein into a highly particular tertiary architecture. Such spatial arrangements alter the chemical environments of certain residues, dramatically expanding their abilities and generating emergent behaviour that could never occur without specific orientation of the constituent amino acids: catalysis, allosteric responsivity, target recognition, etc. Similar behaviour is observed in nucleic acid macromolecules.
Accordingly, understanding the mechanisms and thermodynamics of self-assembly is a powerful tool for progress in many fields: biochemistry, nanotechnology and medicine chief among them. In this regard, proteins are still too complicated and can only be investigated through computationally expensive molecular simulations. Prediction of the three-dimensional structure of proteins ab initio remains a grand challenge. Polyelectrolytes, however, are simpler than polypeptides; generally the number of different monomer species is 1–3 rather than the 20 naturally occurring amino acids, yet polyelectrolytes still retain dynamic behaviour, and their complexation process has parallels to protein folding. While polyelectrolyte self-assembly typically does not rely on detailed information in the sequence of monomers as biomacromolecules do, it is dependent on the same physical principles (electrostatics, bulkiness, hydrophobicity, etc.) and is especially similar in the case of protein misfolding, such as in Alzheimer's and Parkinson's disease (Forloni, 2002). Examination of PEC self-assembly is therefore a useful companion to protein studies and, more importantly, introduces the potential for customisation of PEC architecture de novo via tailoring of the constituent polyelectrolyte chemistry, if self-assembly is sufficiently understood, a directly applicable tool in materials science (Sing, 2017).
The PEC self-assembly process appears somewhat marvellous from a thermodynamic perspective. The Second Law of Thermodynamics dictates that a molecular behaviour occurs spontaneously only if it lowers the Gibbs Energy of the system. Essentially, an action is allowed if it decreases the net instability of all molecules involved (tracked by a variable called enthalpy) or if it increases entropy; the Gibbs Energy simply weighs these factors. Entropy, while difficult to understand conceptually, generally tracks the number of accessible configurations, called microstates, of particles in the system. Consider a messy room: the less organised it is, the more ways specific objects can be moved around without changing the overall appearance and properties of the room. It can be said to have higher entropy than a tidy room, which must retain a certain configuration – or microstate – to be tidy. Polyelectrolyte self-assembly can be observed to sequester dissolved and distributed polymers into a condensed phase apart from the rest of the solution, ordering the system, decreasing entropy and discouraging spontaneous complexation. Intuitively that leaves electrostatic attraction, the obvious interchain stabilising (enthalpic) interaction facilitated by the condensed phase, to be the driving force of complexation.
This assumption, largely accepted for decades, is incorrect (Fu and Schlenoff, 2016). Recent examinations of polyelectrolyte complexation indicate that the enthalpy of complexation accounts for only up to 10 per cent of the Gibbs Energy, and that inter-polymer electrostatics is not primarily responsible even for this small portion (Kayitmazer, 2017). The thermodynamic driving force for this powerful behaviour must lie elsewhere.
The subtlety comes with the counterion species, included with each polyion to balance charge before mixing. These are generally highly soluble small ions, like sodium and chloride. Before oppositely charged polyelectrolytes are mixed, their counterions associate with the chains electrostatically and form a local 'atmosphere' of charge in solution in a process called Manning Condensation, which stabilises the polyelectrolyte chain (Sing, 2017; Manning and Ray, 1998). During complexation, this atmosphere of counterions is replaced by the oppositely charged polyelectrolyte, destabilising the chains as the atmosphere-chain interactions are lost and then re-stabilising with the approach of the opposing polyion. Depending on the characteristics of the counterions and polyelectrolytes, the net effect may be favourable, negligible or even unfavourable (Fu and Schlenoff, 2016; Priftis et al., 2012). Yet complexation is favourable and spontaneous.
The true source of the 10 per cent enthalpic contribution is the solvation of released counterions (Gummel et al., 2007). Because water is highly polar (the electronegativity of the central oxygen atom causes a localised separation of charge across the molecule, with the oxygen becoming slightly negative and the hydrogens slightly positive), water molecules interact with ions by forming solvation shells, loose cages of water molecules around the ion, which maximise the electrostatic ion-dipole stabilisation. Together, the enthalpic contributions of solvation shells in the supernatant easily overwhelm that of electrostatic enthalpy within the sequestered complex phase (Fu and Schlenoff, 2016).
The remaining 90 per cent of the Gibbs Energy driving force for complexation derives from the increase in entropy during release of the counterions from the polyelectrolytes. Because the counterions are localised near their polyelectrolyte chain in the 'atmosphere' before complexation, they are constrained in their movement and position – their entropy is low. Upon complexation, these counterions are freely released into the bulk solution phase and gain significant entropy, drastically altering the Gibbs Energy and assuring spontaneity of self-assembly (Fu and Schlenoff, 2016).
It should be noted that while the enthalpic contribution of interactions within the condensed PEC phase is small compared to the contribution of the released counterions, this does not mean these interactions are negligible. This is especially in the case of more complex polyelectrolyte structure, in which a greater variety of non-covalent intermolecular forces may come into play, interactions may meaningfully affect the structure of the PEC phase. Characteristics of the polymer chain, including rigidity (Lueckheide et al., 2018), hydrophobicity (Sadman et al., 2017), hydrogen-bonding ability (Marciel et al., 2018), can introduce order into the disorganised condensed phase or alter mechanical and dynamic properties, including producing rod-like substructures (Marciel et al., 2018). Distribution of charged monomers along the chain also have a profound effect (Chang et al., 2017). While these secondary interactions are beyond the scope of this perspective, they serve to illustrate the tuneability of the complex phase.
Even before detailed thermodynamic studies of complexation, polyelectrolyte complexes were known to be reversible in high-salinity environments (Krogstad et al., 2014b). The prevailing theory posited that extra free ions (often just counterion salts) could penetrate into the PEC and disrupt the electrostatic associations between polyelectrolytes via a 'charge screening' process. Essentially, by encouraging small, mobile ions to compete with similarly charged polyelectrolytes for binding, the electrostatic interchain bonds could be replaced by free ion-polymer bonds, dissolving the PEC back into free-floating polyelectrolyte chains in condensed counterion atmospheres. This phenomenon can now be rationalised by more complex entropic arguments regarding the microphase-separation equilibrium. The end result is the same: mix oppositely charged polyelectrolytes and a complex will form. Add salt and the complexes disappear again.
Again, polyelectrolyte structures adds nuance to this phase transition. Higher charge-density on the backbone, hydrophobicity and chain lengths reduce polymer mobility and make the complex phase more difficult to dissolve. Often, strong polyelectrolytes form waxy or glassy solid-like PECs when no extra salt is present. Increasing salt concentration first loosens the phase into the liquid-like coacervate state, before completely dissolving it. This spectrum of PEC phases, from hard solids to liquids to a homogenous polymer solution, is termed the 'complex-coacervate continuum' (Wang and Schlenoff, 2014). Interestingly, not every state on the continuum is at stable equilibrium. Solid PECs are often kinetically-trapped due to the strength of inter-polymer attractions and do not reach electroneutrality, equilibrium size or shape (Fu et al., 2017). Thus compositions of the supernatant and complex phase change across the continuum, not just their structure and properties (Spruijt et al., 2010).
Every PEC moves across the continuum differently at different salt concentrations, and salt identity also has a strong effect. The ability of the added salt to disrupt polymer-polymer electrostatics depends on the Pearson hard–soft matching of ions, and also on their ability to interact with the solvation shells of each ionic species – the Hofmeister series (Salomäki and Kankare, 2008; Salomäki et al., 2004). The Hofmeister series ranks common ions by their ability to restructure water. Chaotropic ions like bromide break the hydrogen-bonding network of water around polyions and more effectively dissolve the PEC phase. Cosmotropic ions such as acetate structure water more effectively and strengthen PECs. Added salts thus offer a further mechanism for modulating the properties of PECs beyond static chemical structure.
By using one or more weak polyelectrolyte species that are pH sensitive, a similar effect can be achieved through changes in pH. For example, polyacrylic acid (PAA, a polyanion) has been shown to create PECs at neutral pH that disassemble in 12 hours at pH5 upon protonation (charge neutralisation) of the carboxylate moieties. More advanced chemistries can theoretically be used to induce structural change with many common stimuli, such as certain biomolecule targets, light and temperature (Aluri et al., 2009). PECs thus show potential for generalised stimuli-responsivity; they can be considered 'smart' materials.
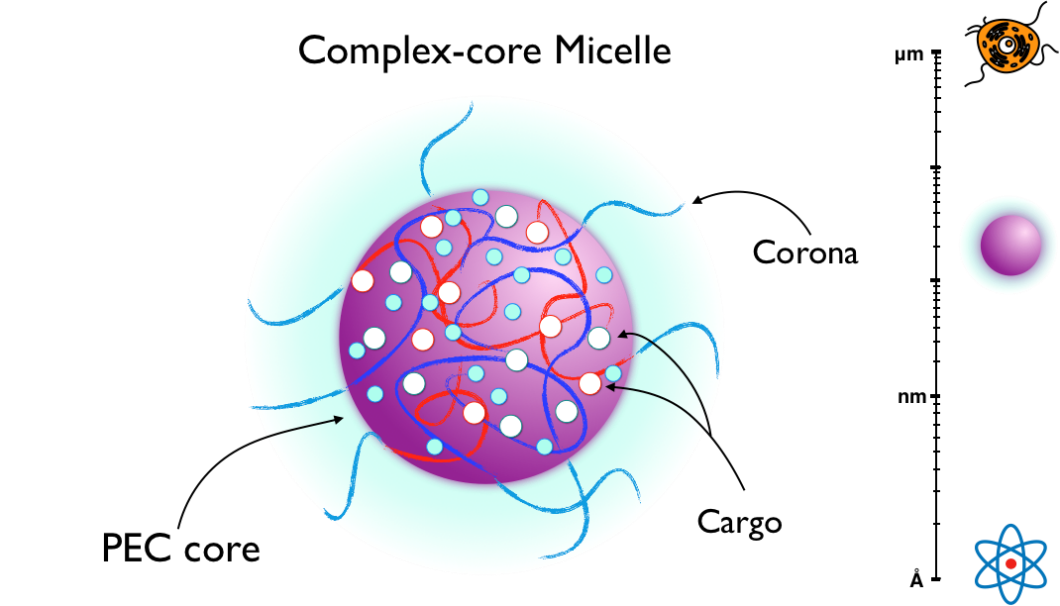
Importantly, these key attributes of PECs – self-assembly and modular stimuli-responsivity – are accessible not just to complexes of synthetic polyelectrolytes but also to complexes involving block copolymers with charged domains, charged biological macromolecules like DNA and other compounds such as pharmaceutical molecules (Voets et al., 2009). PECs thus show exciting activity as customisable drug-delivery vehicles (Acar et al., 2017). Particularly common is the use of diblock copolymers (polymers separated into two domains) incorporating a charge-bearing block and a hydrophilic block such as polyethylene glycol (PEG). By tuning the electrostatic attraction between complexing blocks and neutral block length, PECs can be formed as nanoparticles instead of bulk phases (Van der Kooij et al., 2012). These 'complex-core micelle' nanoparticles, which contain a PEC core (generally a liquid-like coacervate) generated by the charged block, protected by a water-soluble corona from the neutral block, still self-assemble when copolymers are mixed and dissemble in salt. Additional targeting and sensory functionalities, such as grafted antibodies, can be incorporated into the corona block (Voets et al., 2009). Therapeutic molecules can be loaded into the charged core during self-assembly and released in a controlled manner by the stimulated dissociation of the PEC core.
Particularly facile is the incorporation of nucleic acids (DNA and RNA) into the core because these molecules contain a negatively-charged phosphodiester backbone that actively associates with positive charges in the complex core. In many cases, DNA can act as the sole polyanion, forming a stable micelle entirely without the benefit of an additional negatively-charged macromolecule (Voets et al., 2009). Metal-centred pharmaceuticals, such as cisplatin, also interact with Lewis-basic polyanions and are very easily integrated into the micelle (Masayuki et al., 1996). These charge-based interactions highlight a key advantage of PEC complex-core micelles over more traditional amphiphilic micelles (which assemble a hydrophobic core, similar to lipid bilayers), namely the charge-dense PEC core, which remains partially hydrated. The presence of water and charge significantly boosts the stability of pharmaceutical species with a fragile tertiary structure stored in the core, preventing the degradation of nucleic acids and proteins during delivery, a shortcoming which continues to plague amphiphilic micelles (Acar et al., 2017).
If PEC structure-function relations can be properly understood, this technology has particular potential in the treatment of cancer. In theory, solutions of pharmaceuticals and custom polyelectrolytes could be injected near the tumour, where the polyelectrolytes would self-assemble into PEC complex-core micelles in the bloodstream, incorporating therapeutic cargo in the process. The micelles then approach the tumour and are endocytosed, where the micelle can react to any number of cancer-specific stimuli. The low intracellular pH of many cancers, presence of apoptosis suppressor molecules or other uncommon conditions make convenient triggers for PEC reversion and disintegration of the particle. The pharmaceutical is then released at the location of the tumour, killing the cancerous cells. This method minimises many of the most important complications in contemporary cancer treatment, especially the prevalence of dangerous iatrogenic side-effects during chemotherapy. Because the cargo-release stimulus is customisable, micelles that are taken up by healthy cells will not rupture, and will eventually be degraded safely by the lysosome. The PEC micelle effectively provides containment and targeting functionality to chemotherapy drugs, drastically increasing their effectiveness and safety (Acar et al., 2017).
Astute readers by now will have recognised the key features of polyelectrolyte complexes – spontaneous self-assembly by phase-separation, stimuli-responsivity and the ability to sequester biomolecules such as nucleic acids – as useful attributes for components of primitive life forms. To address the possible role of polyelectrolytes in the chemical origin of life, and, through this discussion, the implicit possibility of the creation of synthetic organisms via this technology, we must first characterise minimal chemical life.
The precise definition of minimal chemical life has been debated for decades, and many experts would forego a strict definition altogether in favour of a more general description (Luisi, 1998). While the field is far from settled, most would agree that minimal chemical life should encompass three functionalities which are chemically integrated and co-regulated: a container, a metabolism and a program (Bedau, 2011). These functionalities are defined in their broadest senses, such that a container need only be an apparatus that allows a system to retain its identity over time in contrast to its environment, a metabolism is any means of harvesting energy and materials from the environment and a program is any heritable and changeable information that governs the structure or activity of the other two components.
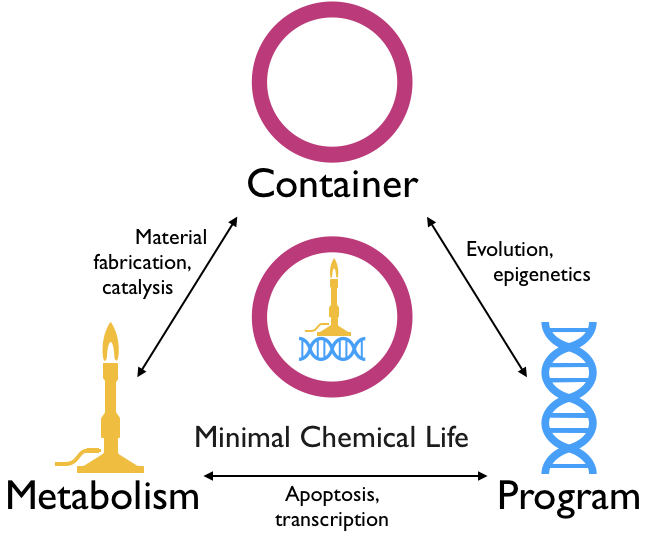
Equally important is that the container, program and metabolism are not merely spatially proximate but chemically interrelated, such that changes in one affect the activity of the others (Szostak et al., 2001). For example, the container may concentrate or organise internal chemical species so as to catalyse metabolism, the metabolism may produce materials for incorporation into the container and program, and the program may alter the structure and function of the container and metabolic components based on stimuli or random variation. These systematic interdependencies eventually allow the emergence of dynamic behaviours that more recognisably characterise life, such as evolution, reproduction and stress responses (Bedau, 2011).
Polyelectrolyte complexes could therefore fill the role of a prebiotic container that might house metabolic and programmatic apparatus. A polyelectrolyte-based theory of the origin of life was posited in the 1930s by Alexandrei Oparin, a Soviet biochemist (Oparin, 1953). However, due to the perceived implausibility of polyions on the prebiotic Earth, this theory was largely discredited until recently, when it was shown to solve many problems in the dominant amphiphilic lipid-membrane model (McNichol, 2008).
Polyelectrolyte theory fittingly takes advantage of coacervate phase-separation to divide space into in and out. Unlike neutral polymer species, which require high concentrations to gel into a sheltered material, the substantial negative Gibbs Energy for polyelectrolyte complexation allows microphase-separation to occur at relatively low thresholds, which would have been the norm in the prebiotic ocean of the early Earth (Keating, 2012). As has been previously mentioned, thanks to its highly charged interior, this polyelectrolyte phase can sequester useful organic and inorganic compounds with surprising efficiency: in one study, complexes of poly(lysine) (PLys, a polymerised amino acid bearing positive charges) and negatively-charged adenosine triphosphate (ATP) encapsulated porphyrins, metal-oxide nanoparticle oxidisers, organic and inorganic dyes, and simple enzymes (Koga et al., 2011). These ancient molecules help facilitate important biological reactions, including signalling, protein-building and photosynthesis, and were extracted from the surrounding solution into the PEC with efficiencies, in some cases over 99.95 per cent. The coacervate droplets were stable in a relatively wide pH range, reversible outside of it and difficult to permanently degrade. Moreover, the interior of the PLys–ATP complex formed α-helical strands spontaneously, motifs similar to the ubiquitous protein domain. This secondary structure could endure dephosphorylation to PLys–ADP by enzymes contained within its own volume without loss of compartmentalisation or phase integrity in solution, thus proving durability during cellular metabolic transformations even without an external membrane layer. The authors note that these catalytic transformations were twice as fast in the PEC droplets compared to similar conditions in the supernatant, not just because of the high concentration of enzyme sequestered but also because of mediation from the highly ionic coacervate environment (Koga et al., 2011). Such an example demonstrates the ability of polyelectrolytes to complex from low concentration, gather useful materials spontaneously, and retain compartmentalisation of these materials away from the environment during advanced chemical processes required for familiar life forms.
The PEC droplets mentioned above operated as proto-cells without a sheltering membrane. However, neutral macromolecules and amphipaths such as lipids have been shown to accumulate on the interface between coacervate and aqueous phases through the hydrophobic effect, similar to the entropic driving force behind complexation (Keating, 2012). Water molecules surrounding the PEC phase and non-hydrophillic neutral species in the bulk solution are highly ordered in their orientation because of the lack of hydrogen bonding from the non-aqueous molecules. Thus any interface between water and these hydrophobic molecules decreases entropy and is unfavourable. The configuration with lipids coating the PEC phase minimises this interface, releasing ordered water into the bulk instead of condensed counterions as in PEC complexation (Helfrich et al., 2002). This action also creates a formal semipermeable membrane which stabilises the PEC.
Once a membrane such as a lipid layer forms, PEC phase-separation acts as a chemical handle for further control of complexity in and around the membrane. Because macromolecules cannot pass through the nascent membrane layer, the quantity of these solutes in the interior of the micelle is fixed. Hence by varying concentration (tonicity) in the surrounding solution, water can be pushed into or out of the membrane compartment by osmosis, changing the interior volume of the vesicle and the hydration of the polymer phase. However, if enough water enters into the membrane compartment, the interior hydrated coacervate will phase-separate a second time into aqueous and PEC phases. Addition of lipid material into the system will then increase the surface area (but not volume) of the membranes and encourage budding – where the membrane splits between the two interior phases, creating inhomogeneous daughter vesicles. Solutes that are partitioned asymmetrically across phases will be inherited asymmetrically as well, maintaining the partitioning of key species in the proto-cells (Keating, 2012).
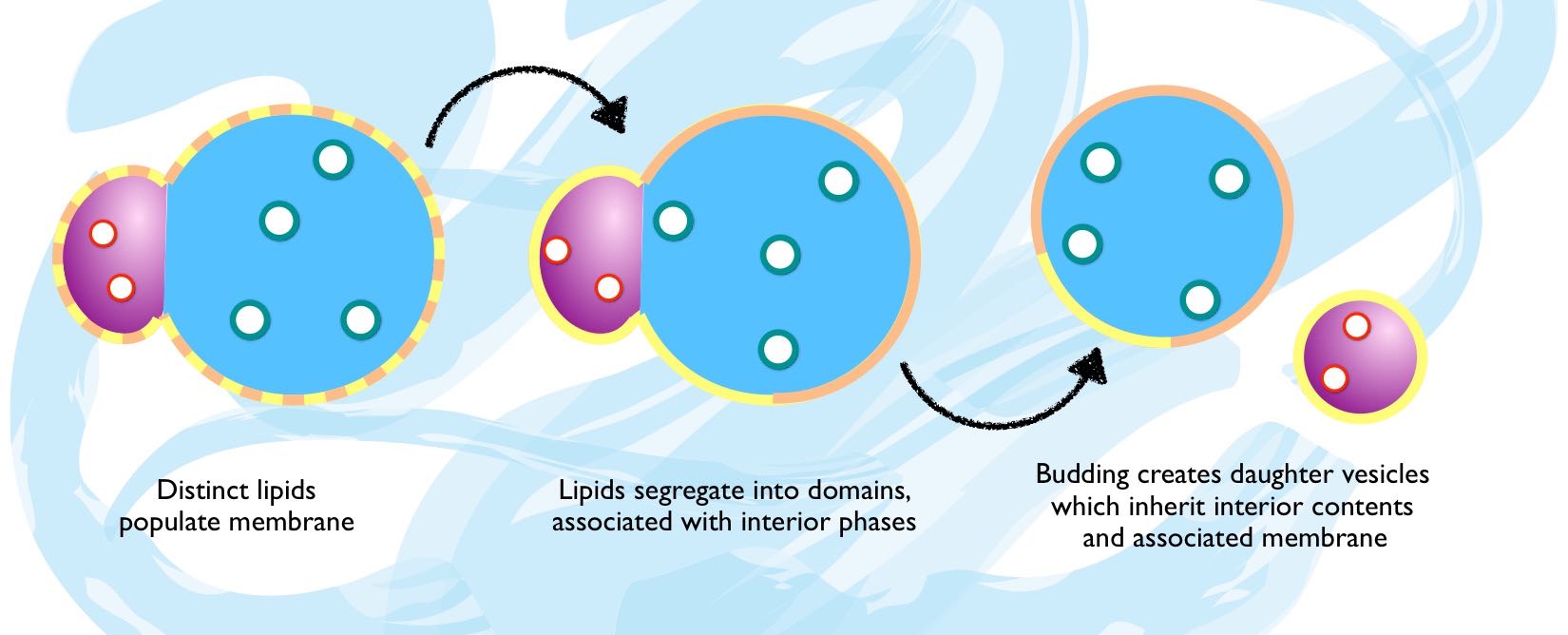
However, different species of lipid often co-accumulate in a membrane via similar principles to PEC complexation and organise into domains – two-dimensional phases of different lipids and associated membrane molecules (i.e. proteins). If this should occur, the lipid phases will interact with interior phases with different affinities, and so certain lipid-membrane domains will spontaneously become associated with polyelectrolyte or aqueous interior phases and their accompanying solutes as a result of more thermodynamic interactions between the membrane and interior. When budding, these 'pinned' lipid-polymer phases segregate together, allowing for full and robust differentiation of the protocell surface, interior environment and contents. Accordingly, the organisation of volume through PEC phase-separation begets the organisation of surface material and functionality, as well as additional complexity. This is a necessary condition for the evolution of transmembrane protein complexes for sensing, communication, locomotion and many other advanced cellular processes (Keating, 2012).
Polyelectrolyte complex coacervation thus constitutes a primitive cellular container, interacting with many plausible programmatic and metabolic building blocks, and templates the formation of highly complicated anisotropic cellular membranes. Intricacy arises spontaneously as a result of the complexation-driven phase-separation, given the correct polyelectrolytes in solution, and in doing so, supplies many non-trivial prerequisites for advanced life forms: robustness, responsivity and spontaneous organisation. Applications for such technology include nanoscale reaction vessels that can utilise the extreme control of local conditions such as ionic strength and concentration to safely access chemical pathways normally only possible in exceptionally harsh and toxic industrial processing. Eventually, PECs might also house attempts at artificial cellular life.
Polyelectrolyte complexes are a sophisticated and dynamic class of materials relevant to numerous fields concerning living systems. These phases are created through an entropy-driven self-assembly process via subtle interactions with counterions and aqueous solvents, accessing a wide spectrum of states from solid-like to liquid-like. Incorporation of charged polymer domains in block copolymers facilitates the assembly of complex-core micelle nanoparticles with unusual tuneability. These particles are thus extremely promising drug-delivery vehicles, with the opportunity for extensive customisation of release trigger, targeting functionality and structural characteristics. Furthermore, the fundamental ability of polyelectrolyte complexes to spontaneously create and maintain physical complexity at low concentrations in simple solutions makes them intriguing model systems for investigations into the origins of chemical life and the future creation of artificial life.
The far-reaching applications of PECs and their derivatives are linked by their dependence on the fundamental attribute of these complexes: spontaneous and reversible organisation of space and matter. While the ability to create chemical and spatial order under certain conditions is not unique to PECs, few other technologies possess it with such robustness and versatility under benign conditions. This feat is intimately related to local entropy changes, and thus the subtle thermodynamic interactions of polyelectrolyte complexation must be well understood in order to expand our ability to manipulate action on the nanoscale, and develop the technologies that arise from it.
The author gratefully acknowledges fruitful discussions with J. T. and J. C.
All images are the author's own
Acar, H., J. M. Ting, S. Srivastava, J. L. LaBelle and M. V. Tirrell (2017), 'Molecular engineering solutions for therapeutic peptide delivery', Chem. Soc. Rev., 46, 6553–69
Aluri, S., S. M. Janib and J. A. Mackay (2009), 'Environmentally responsive peptides as anticancer drug carriers', Adv. Drug Deliver. Rev., 61, 940–52 doi:10.1016/j.addr.2009.07.002
Balchin, D., M. Hayr-Hartl and F. U. Hartl (2016), 'In vivo aspects of protein folding and quality control', Science, 353, 6294 doi:10.1126/science.aac4354
Bedau, M. A. (2011), 'A functional account of degrees of minimal chemical life', Synthese, 185, 73–88 doi:10.1007/s11229-011-9876-x
Chang, L. W., T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez, C. E. Sing and S. L. Perry (2017), 'Sequence and entropy-based control of complex coacervates', Nat. Commun., 8, 1–7
Forloni, G., L. Terreni, I. Bertani, S. Fogliarino, R. Invernizzi, A. Assini, G. Ribizzi, A. Negro, E. Calabrese, M. A. Volonté, C. Mariani, M. Franceschi, M. Tabaton, A. Bertoli (2002), 'Protein misfolding in Alzheimer's and Parkinson's disease: Genetics and molecular mechanisms' Neurobiol. Aging. doi:10.1016/S0197-4580(02)00076-3
Fu, J. andJ. B. Schlenoff (2016), 'Driving forces for oppositely charged polyion association in aqueous solutions: Enthalpic, entropic, but not electrostatic', J. Am. Chem. Soc., 138, 980–90 doi:10.1021/jacs.5b11878
Fu, J., H. M. Fares and J. B. Schlenoff (2017), 'Ion-pairing strength in polyelectrolyte complexes', Macromolecules, 50, 1066–74 doi:10.1021/acs.macromol.6b02445
Gummel, J., F. Cousin and F. Boué (2007), 'Counterions release from electrostatic complexes of polyelectrolytes and proteins of opposite charge: A direct measurement', J. Am. Chem. Soc., 129, 5806–07 doi:10.1021/ja070414t
Guvendiren, M., H. Lu and J. Burdick (2012), 'Shear-thinning hydrogels for biomedical applications', Soft Matter, 8, 260−72
Helfrich, M. R., L. K. Mangeney-Slavin, M. S. Long, K. Y. Djoko and C. D. Keating (2002), 'Aqueous phase separation in giant vesicles', J. Am. Chem. Soc., 124, 13374–75 doi:10.1021/ja028157
Kayitmazer, A. B. (2017), 'Thermodynamics of complex coacervation', Adv. Colloid Interf.., 239, 169–77 doi:10.1016/j.cis.2016.07.006
Keating, C. D. (2012), 'Aqueous phase separation as a possible route to compartmentalization of biological molecules', Accounts Chem. Res., 45, 2114–24 doi:10.1021/ar200294y
Koga, S., D. S. Williams, A. W. Perriman and S. Mann (2011), 'Peptide-nucleotide microdroplets as a step towards a membrane-free protocell model', Nat. Chem., 3, 720–24 doi:10.1038/nchem.1110
Krogstad, D. V., N. A. Lynd, S. H. Choi, J. M. Spruell, C. J Hawker, E. J. Kramer and M. V. Tirrell (2014a), 'Effects of polymer and salt concentration on the structure and properties of triblock copolymer coacervate hydrogels', Macromolecules, 46, 1512–18 doi:10.1021/ma302299r
Krogstad, D. V., N. A Lynd, D. Miyajima, J. Gopez, C. J. Hawker, E. J. Kramer and M. V. Tirrell (2014b), 'Structural evolution of polyelectrolyte complex core micelles and ordered-phase bulk materials', Macromolecules, 47, 8026–32 doi:10.1021/ma5017852
Li, L., S. Srivastava, M. Andreev, A. B. Marciel, J. J. De Pablo and M. V. Tirrell (2018), 'Phase behavior and salt partitioning in polyelectrolyte complex coacervates', Macromolecules, 51, 2988–95
Lueckheide, M., J. R. Vieregg, A. J. Bologna, L. Leon and M. V. Tirrell (2018), 'Structure-property relationships of oligonucleotide polyelectrolyte complex micelles', Nano Lett.. 18, 7111–17 doi:10.1021/acs.nanolett.8b03132
Luisi, P. L. (1998), 'About various definitions of life', Origins Life Evol. B., 28, 613–22 doi:10.1023/a:1006517315105
Manning, G. S. and J. Ray (1998), 'Counterion condensation revisited'. J. Biomol. Struct. Dyn., 16, 461–76 doi:10.1080/07391102.1998.10508261
Marciel, A. B., S. Srivastava and M. V. Tirrell (2018), 'Structure and rheology of polyelectrolyte complex coacervates', Soft Matter, 14, 2454–64
Masayuki, Y., T. Okano, Y. Sakurai, S. Suwa and K. Kataoka (1996), 'Introduction of cisplatin into polymeric micelle', J. Control Release, 39, 351–56 doi:10.1016/0168-3659(95)00165-4
McNichol, J. (2008), 'Primordial Soup, Fool's Gold andSpontaneous Generation', BIOCHEM EXP BIOL, 36, 255–61 doi:10.1002/bmb.20194
Oparin, A. I. (1953), The Origin of Life, Mineola, NY: Dover Publications
Overbeek, J. T. G. and M. J. Voorn (2005), 'Phase separation in polyelectrolyte solutions. Theory of complex coacervation'. J. Cell. Comp. Physiol. doi:10.1002/jcp.1030490404
Priftis, D., N. Laugel, and M. V. Tirrell (2012), 'Thermodynamic characterization of polypeptide complex coacervation', Langmuir. 28, 15947–57 doi:10.1021/la302729r
Ramasamy, T., B. K. Poudel, H. Ruttalaa, J. Y. Choi, T. D. Hieua, K. Umadevi, Y. S. Younc, H. G. Choi, C. S. Yonga and J. O. Kima (2016), 'Cationic drug-based self-assembled polyelectrolyte complex micelles: Physicochemical, pharmacokinetic andanticancer activity analysis', Colloid Surface B, 146, 152–60 doi:10.1016/j.colsurfb.2016.06.004
Sadman, K., Q. Wang, Y. Chen, B. Keshavarz, Z. Jiang, K. R. Shull (2017), 'Influence of hydrophobicity on polyelectrolyte complexation', Macromolecules, 50, 9417–26, doi:10.1021/acs.macromol.7b02031
Salomäki, M. and J. Kankare, (2008), 'Specific anion effect in swelling of polyelectrolyte multilayers', Macromolecules, 41, 4423–28 doi:10.1021/ma800315j
Salomäki, M., P. Tervasmäki, S. Areva and J. Kankare (2004), 'The Hofmeister anion effect and the growth of polyelectrolyte multilayers', Langmuir., 20, 3679–83 doi:10.1021/la036328y
Sing, C. E. (2017), 'Development of the modern theory of polymeric complex coacervation', Adv. Colloid Interfac., 239, 2–16 doi:10.1016/j.cis.2016.04.004
Spruijt, E., A. H. Westphal, J. W. Borst, M. A. Cohen Stuart and J. Van Der Gucht (2010), 'Binodal compositions of polyelectrolyte complexes', Macromolecules 43, 6476–84
Szostak, J. W., D. P. Bartel and P. L. Luisi (2001), 'Synthesizing life', Nature, 409, 387–90 doi:10.1038/35053176
Tekaat, M., D. Bütergerds, M. Schönhoff, A. Fery and C. Cramer (2015), 'Scaling properties of the shear modulus of polyelectrolyte complex coacervates: A time-pH superposition principle', Phys. Chem. Chem. Phys., 17, 22552–56 doi:10.1039/c5cp02940f
Ting, J. M., H. Wu, A. Herzog-Arbeitman, S. Srivastava and M. V. Tirrell (2018), 'Synthesis and assembly of designer block polyelectrolytes', ACS Macro Lett., 7, (6), 726–33
Van Der Kooij, H. M., E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. Van Der Gucht (2012), 'On the stability and morphology of complex coacervate core micelles: From spherical to wormlike micelles'. Langmuir, 28, 14180–91
Voets, I. K., A. de Keyser and M. A. Cohen Stuart (2009), 'Complex coacervate core micelles', Adv. Colloid Interface., 147–148, 300–18 doi:10.1016/j.cis.2008.09.012
Wang, Q. and J. B. Schlenoff (2014), 'The polyelectrolyte complex/coacervate continuum', Macromolecules., 47, 3108–16 doi:10.1021/ma500500q
Zhao, Q., D. W. Lee, B. K. Ahn, S. Seo, Y. Kaufman, J. N. Israelachvili and J. H. Waite (2016), 'Underwater contact adhesion and microarchitecture in polyelectrolyte complexes actuated by solvent exchange', Nat. Mater., 15, 407–12 doi:10.1038/nmat4539
Zhang, H., C. Wang, G. Zhu and N. S. Zacharia (2016), 'Self-healing of bulk polyelectrolyte complex material as a function of pH and salt', ACS Appl. Mater. Interfaces, 8, 26258–65 doi:10.1021/acsami.6b06776
To cite this paper please use the following details: Herzog-Arbeitman, A. (2019), 'Polyelectrolyte Complexation: Entropy, medicine and the beginning of life', Reinvention: an International Journal of Undergraduate Research, Volume 12, Issue 2, https://reinventionjournal.org/article/view/338. Date accessed [insert date]. If you cite this article or use it in any teaching or other related activities please let us know by e-mailing us at Reinventionjournal@warwick.ac.uk.